Molecular Magnets
In this project magnetic properties of magnetic molecules are investigated. Also, different sample substrate systems with different electronic and magnetic properties were chosen to reveal the coupling of the molecules to each other and to the substrate. In order to leave the electronic structure of an adsorbed molecule mostly unchanged and to observe it in its original state, atomic force microscopy is deployed, since this method is not limited to conductive surfaces. As insulating substrates sodium chloride (NaCl - nonmagnetic) and nickel oxide (NiO - antiferromagnetic at (001) surface) are used in this experiment. The third substrate system which also serves as a candidate for a magnetic coupling with magnetic molecules is the iron monolayer on tungsten (W(001)) 1. Former magnetic exchange force microscope (MExFM) studies were performed on this system as well as on nickel oxide and the antiferromagnetic order could be reproduced in both cases2,3. A possible magnetic coupling from the substrate to the metallic center of a molecule is investigated. For that reason different magnetic molecules, which exhibit at least one metallic center, are used (see figure 1 for structural formula).

Figure 1: Structural formula of a Co-Salen (C16H14CoN2O2) and b CuNi-Salen (C18H14CuN2NiO7) or Doublesalen.
Further magnetic investigation of single molecules or molecular nanostructures only is possible if one is aware of the interactions between the molecules and the molecule substrate interaction. Therefore growth studies are inevitable. Figure 2 is showing the result of the study of Co-Salen on NaCl(001)4. In fig. 2.a compact nanocrystallites (red) and metastable nanowires (blue) are coexisting at the same time. Structural resolution shows the well-ordered molecular unit cell consisting out of dimers in 2.b,c and the atomic lattice of NaCl(001) in 2.d. The compact nanocrystallites are thermodynamically favored, meaning that post annealing or increasing the elapsed time results in dissolving nanowires – at a certain point only nanocrystallites are left on the NaCl(001) surface. The metastable bimodal growth mode at hand reveals a rather weak molecule substrate interaction and high molecule molecule interaction.

Figure 2: a 1 x 1 µm² of 5 monolayer Co-Salen on NaCl(001) with two crystalline formes of Co-Salen (red and blue) b, c, d molecular/atomic resolution of the structures given in a.
If one changes the substrate to NiO, not only the magnetic properties are altered, but also the electronic structure, for that reason the growth is quite different compared to Co-Salen on NaCl. Figure 3 shows the result of the growth study of Co-Salen on NiO(001). By depositing a small amount of molecules to the surface, stepedge decoration is observed (fig 3.a). But until the coverage reaches a monolayer no growth occurs: The molecules diffuse rather freely above the surface terraces, without forming any nucleation (fig 3.b). If the density reaches a monolayer, a closed disordered wettinglayer emerges. Thereupon, further growth continous with higher molecular density (fig 3.c). Showing no crystalline growth signifies a higher molecule substrate interaction, than molecule molecule interaction. A possible explanation is the known oxygen affinity of Co-Salen5, which might bind coordinativly with an oxygen atom of the NiO and therefore could cause a higher interaction.

Figure 3: a At low coverages step edge decoration starts until all step edges are fully occupied (80 x 80 nm²). b Increasing the coverage leads to diffusing molecules at the terraces, which is revealed due to stripes in the topogrophy data (80 x 80 nm²). c At higher coverages (5 ML) a crystalline growth of the Co-Salen forms a lacerated layer growth with the amorphous monolayer beneath it (2 x 2 µm²).
To understand and control magnetism of molecules one has to evaluate the adsorption geometry of the molecules. For that reason the adsorption site and orientation was determined experimentally and theoretically approved for Co-Salen6. Knowing that the interaction above an anion of an ionic insulator to a metallic tip is always largest7, we could unambiguously assign the depressions and protrusions in the atomically resolved images to the respective ionic species and identify the adsorption site of the central Co atom of the Co-Salen molecule to be the chloride ion on the NaCl surface and oxygen on the NiO surface (fig. 4).

Figure 4: a Adsorption site of Co-Salen on NaCl(001) (4x4 nm²). b Adsorption site of Co-Salen on NiO(001) (8x8 nm²).
As it is already mentioned above, the NiO(001) surface exihbits an antiferromagnetic aligment of the nickel atoms. Due to a superexchange along the oxygen atoms the nickelatoms are antiferromagnetically coupled – the magnetic moments are parallel to the [111] axis. Since the Co-Salen is adsorbed above the oxygen atom, the superexchange might also lead to a magnetic coupling of the central Co atom with the Ni atoms. The result would be a stabilized out of plane magnetic moment of the Co-Salen center, which one is able to quantify by magnetic exchange force microscopy (MExFM) and magnetic exchange force spectroscopy (MExFS).
Figure 5: Animation of Co-Salen on NiO(001) with two magnetical out of plane moments (up = green, down = red).
Spin-resolved scanning tunneling microscopy on Molecules
Organic as well as magnetic molecules recently attracted growing interest in scientific research, especially in the field of molecular spintronics. Organic molecules are believed to be an advantageous material for transporting the spin degree of freedom, because they are built up of light elements which lead to a low spin orbit coupling and spin relaxation times in excess of 10 ms were already observed in the 1970's [8]. A long spin diffusion length was observed in early spin valve experiments [9,10] but the origin of the measured giant magneto resistance in these experiments was discussed controversially [11]. In particular the morphology of the organic film, e.g. the possibility of diffusion of ferromagnetic material into the organic layer and the role of the organic-ferromagnet interface motivated additional research [12,13]. Spin-polarized scanning tunneling microscopy (SP-STM) is the ideal tool to study the energy and spin distribution of single molecules in atomically well-defined environments on a sub-molecular scale. Thereby, it enables characterization of a well-defined molecule-ferromagnet interface. Furthermore, the comparison with state-of-the-art first principles calculations, including van der Waals interactions, enables unprecedented insight into the physical processes determining the transport of the molecule-ferromagnet interfaces.
To illustrate the capability of SP-STM to study the magnetic properties of organic (and magnetic) molecules, we present here two systems, which have been studied recently in the subgroup “magnetic molecules”. The first example, Co-phthalocyanine molecules on 2 ML of Fe on W(110), shows the spin dependence of the tunneling current flow of molecule-ferromagnet hybrid states. The second example, bis(phthalocyaninato)terbium(III) on Co on Ir(111) demonstrates the ability of SP-STM to determine and map, in real space, the spin splitting of a ligand orbital of a single molecular magnet (SMM).
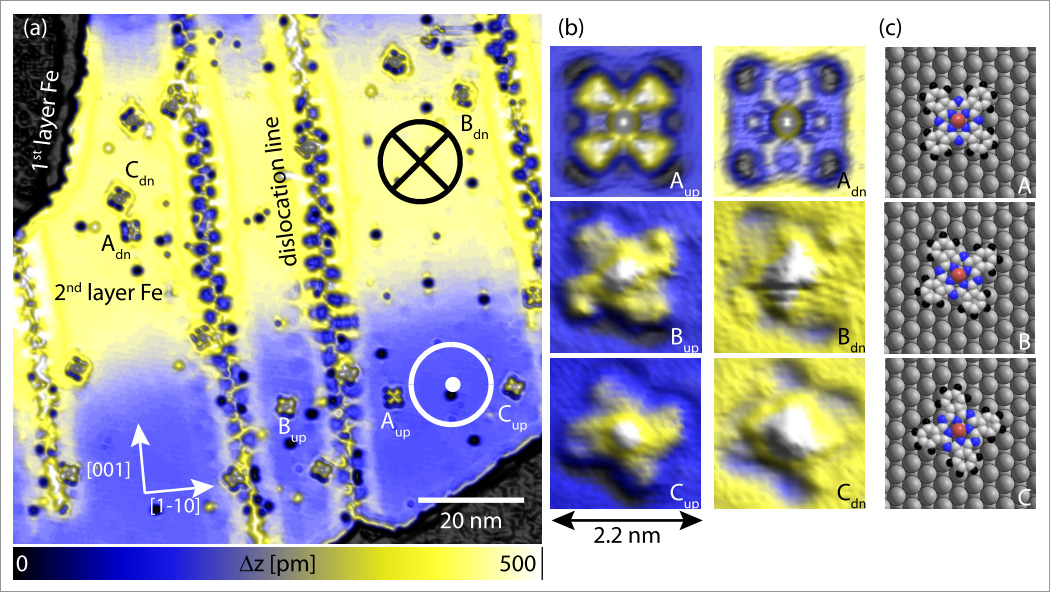
Figure 1: SP-STM topograph of cobalt phthalocyanine molecules (CoPc) on 2 ML of Fe on W110. (a) A spin-resolved overview image shows distinct height variation (from blue to yellow) due to the TMR effect within the atomically flat 2nd layer Fe film. Thereby, the spin-up and the spin-down domain of the sample can be identified. Moreover, individual CoPc molecules can be identified and how a spin-dependent contrast, that can best be seen in the high resolution SP-STM topographs (b): There are three different molecular orientations with respect to the underlying atomic lattice (as schematically depicted in (c) ). All three molecular orientations can be found on both magnetic domains of the Fe. Imaging parameter: I=0.2 nA U= 50 mV
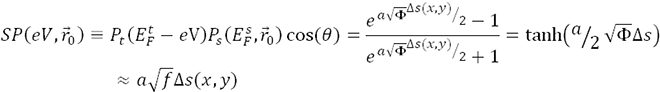
The effective integrated local spin polarization (SP) is determined by the projection of the polarization of the sample (PS) onto the polarization of the tip (PT ). The polarization is given by the normalized difference between the energy integrated density of up-states and the energy integrated density of down states. Here is the interval of integration defined by the Fermi Energy (EF) and the applied bias voltage (eV) [EF, EF +eV]. The constant (a) is approximately 1 eV-1/2Å-1, Φ is the mean local tunneling barrier height, and 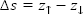 is the relative apparent height difference measured between the spin-up domain (
is the relative apparent height difference measured between the spin-up domain (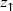 ) and the spin-down (
) and the spin-down (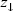 ) domain. It is important to note that the effective integrated local spin polarization is determined at the position of the tip (
) domain. It is important to note that the effective integrated local spin polarization is determined at the position of the tip (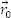 ). Thereby, only sample states which have a slow decay into the vacuum, e.g. pz or dz type states contribute significantly to the measured SP.
). Thereby, only sample states which have a slow decay into the vacuum, e.g. pz or dz type states contribute significantly to the measured SP.
The method as well as the resulting SP map for a CoPc molecule in orientation A adsorbed on the 2nd layer of Fe on W(110) are illustrated in Figure 2. The SP map shows how the spin polarization for the given energy interval [EF, 50meV], measured at the position of the tip, varies on a sub-molecular scale. In particular it demonstrates that electrons originating from the Fe film are primarily of spin-down character, while electrons from the organic ligand of the CoPc are primarily of spin-up character. Thereby, an inversion of the SP at the organic ligand with respect to the underlying Fe-film is clearly resolved.
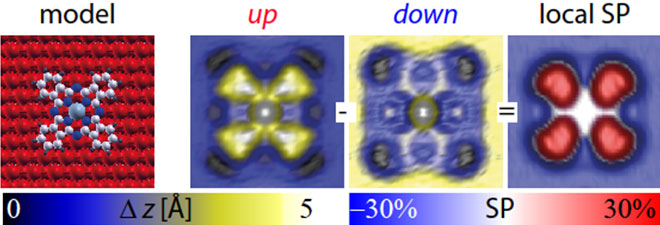
Figure 2: SP-STM topograph of cobalt phthalocyanine molecule in orientation A on 2 ML of Fe on W110. The model geometry (left image), SP-STM topograph of CoPC in the spin-up case (left center image), SP-STM topograph of CoPC in the spin-down case (right center image) and the derived map of the local effective spin polarization (right image).
The origin of this inversion of the SP was determined by comparison with density functional theory simulations (including a correction for van der Waals forces): new metal-organic hybrid states are formed due to the bonding of molecular π orbitals and metal d-bands, due to symmetry considerations these states have high weight in the spin-up density of the states close to the Fermi energy (while the Fe-film has high weight in the spin-down density of states at these energies). The accuracy of the theoretical approach was determined by simulating SP-STM images for the CoPc molecule on both magnetic domains (spin-up and spin-down) as well as simulating SP maps. In order to qualitatively compare theory and experiment, we look at the normalized SP, i.e. we divide the SP of the molecule by the SP of the Fe-film, thereby eliminating (in a first approximation) the influence of the tip polarization (which is not explicitly accounted for in the simulations). Figure 3 shows the convincing agreement between theory and experiment.
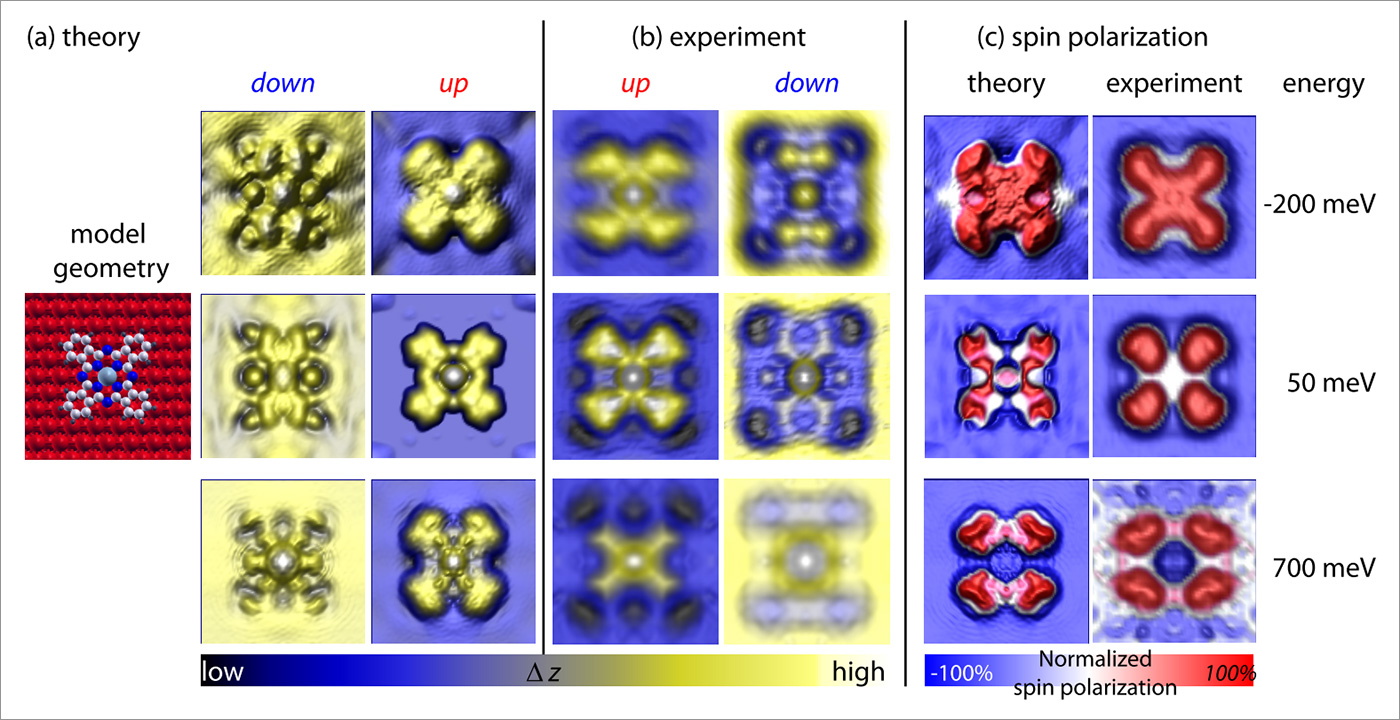
Figure 3: Comparison of simulations based on state-of-the-art first principles calculations and experimental SP-STM images of cobalt phthalocyanine molecules on an ultra-thin Fe film. (a) Simulated SP-STM images for the different spin channels at energies as indicated. (b) experimental SPSTM images. (c) comparison of the experimental and theoretical distribution of the integrated local effective spin polarization.
Moreover, many organic molecules such as the macrocyclic phthalocyanine (Pc) can be functionalized by various metal atoms, to become paramagnetic or superparamagnetic, that is to act as single-molecule magnets. These molecules may be, if anchored on an appropriate surface, used to store, read, or manipulate magnetic information. Crucial toward the end of realizing functional single molecule devices is the understanding how different molecular constituents couple to their surroundings, i.e. how the coupling of the molecule to an electrode modifies intrinsic molecular properties such as charge state, molecular orbital contribution and spin polarization. In recent measurements we were able to resolve the spin splitting of a ligand orbital of a SMM [ bis(phthalocyaninato)terbium(III), abbreviated TbPc2, anchored to a ferromagnetic Co surface. The magnitude of the spin splitting was determined through SP-STS to be in the order of ~210 meV and it is unambiguously shown that both spin-polarized spectroscopic features stem from the same molecular orbital. By taking maps of the local effective spin polarization at energies corresponding to the energy of the spin-resolved spectroscopic features it is shown that both features have the same charge distribution, which is typical for the lowest unoccupied orbital of TbPc2 molecules, but they show opposite spin character (see Fig 4 for an illustration).
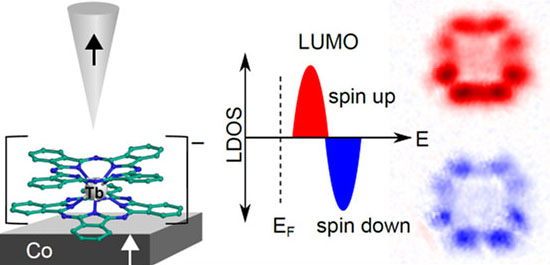
Figure 4: SP-STM on anionic TbPc2 on nanoscale Co islands. Using SP-STM and SP-STS it was possible to measure the spin-splitting of a molecular orbital for the first time in real space. Mapping the charge and spin distribution clearly shows that the same orbital is probed, first in the spin-up channel, than, at higher energies, in the spin-down channel. The magnitude of the splitting is determined to be in the order of ~210 meV, by SP-STS and is induced by the exchange-coupling of the lower PC ring to the Co support.